Sebastian Weber writes:
Bayesian inference has to overcome tough computational challenges and thanks to Stan we now have a scalable MCMC sampler available. For a Stan model running NUTS, the computational cost is dominated by gradient calculations of the model log-density as a function of the parameters. While NUTS is scalable to huge parameter spaces, this scalability becomes more of a theoretical one as the computational cost explodes. Models which involve ordinary differential equations (ODE) are such an example, where the runtimes can be of the order of days.
The obvious speedup when using Stan is to run multiple chains at the same time on different computer cores. However, this cannot reduce the total runtime per chain, which requires within-chain parallelization.
Hence, a viable approach is to parallelize the gradient calculation within a chain. As many Bayesian models facilitate hierarchical models over groupings we can often calculate contributions to the log-likelihood separately for each of these groups.
Therefore, the concept of an embarrassingly parallel program can be applied in this setting, i.e. one can calculate these independent work chunks on separate CPU cores and then collect the results.
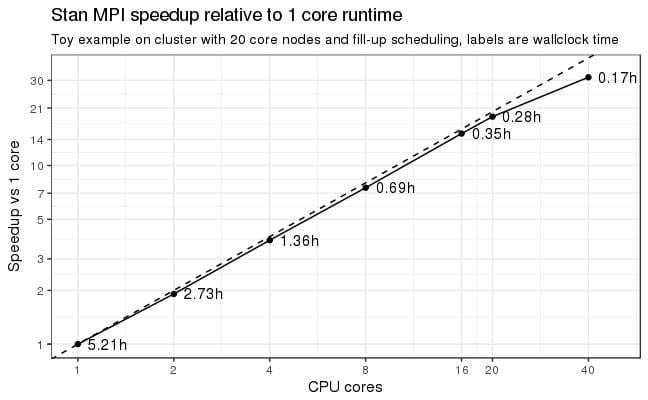
For reasons implied by Stan’s internals (the gradient calculation must not run in a threaded program) we are restricted in applicable techniques. One possibility is the Message Passing Interface (MPI) which spawns multiple CPU cores by firing off independent processes. A root process will send packets of work (sets of parameters) to the child nodes which do the work and then send back the results (function return values and the gradients). A first toy example shows dramatic speedups (3 ODEs, 7 parameters). That is, when going from 1 core runtime of 5.2h we can crank it down to just 17 minutes by using 20 cores (18x speedup) on a single machine with 20 cores. MPI scales also across machines and when throwing 40 cores at the problem we are down to 10 minutes which is “only” a 31x speedup (see the above plot).
Of course, the MPI approach works best on clusters with many CPU
cores. Overall, this is fantastic news for big models as this opens the door to scale out large problems onto clusters which are available nowadays in many research facilities.The source code for this prototype is on our github repository. This code should be regarded as working research code and we are currently working on bringing this feature into the main Stan distribution.
Wow. This is a big deal. There are lots of problems where this method will be useful.
P.S. What’s with the weird y-axis labels on that graph? I think it would work better to just go 1, 2, 4, 8, 16, 32 on both axes. I like the wall-time markings on the line, though; that helped me follow what was going on.
Very cool! I’d be curious to learn what feature of Stan’s internals prevent a shared memory approach. For single node performance (where I suspect most of these codes will run), it seems like OpenMP would be the optimal parallelization scheme. But MPI allows you to port to more machines (particularly to multi-node clusters), so it’s usually a good choice.
It would also be nice to see some weak scaling results, allowing the size of the dataset to grow. That would give a sense of how large a problem one could scale to before the communication dominates.
A great development. I also look forward to some of the work that is being done with Stan on the GPU: http://discourse.mc-stan.org/t/stan-on-the-gpu/326
The coolest thing is that we should be able to combine GPU magic with MPI magic.
Wow, this would be be awesome.
It would be great to hear from the developers what this might look like for a user — would this be ‘plug-and-play’, or would there be aspects that would need to be customized to each model?
Looks like it works for a class of models that fit their parameter grouping criteria. Once in that class, seems like their prototype is pretty plug and play. Based on inspecting this line below
time mpirun -np 4 ./oral_2cmt_mpi sample num_samples=20 num_warmup=150 save_warmup=0 algorithm=hmc stepsize=0.01 adapt delta=0.8 id=$CHAIN data file=mpi_test_parallel4.R init=mpi_init.R random seed=12 output refresh=1 file=samples-$CHAIN-mpi.csv
It’s not quite plug-and-play in the sense of automating this parallelization. We need the parameters and data in a certain format such that we can distribute the work chunks to the child nodes accordingly. In short, you will have to shape your data and parameters in so-called ragged arrays and then we can take it from there. At least that is the current plan.
Sebastian,
I wonder if you could expand on the class of models that would benefit from parallelization. You write “As many Bayesian models facilitate hierarchical models over groupings we can often calculate contributions to the log-likelihood separately for each of these groups.” Do you mean that any model with hierarchical parameters will benefit? Such that, say, y_i = \alpha + \beta * x_i can’t be parallelized, but y_i = \alpha_g[i] + \beta * X would?
I think for hierarchical models the benefit will be the biggest, because then a subset of parameters will be used to evaluate the likelihood of some subset of the data. So you have a natural packing of your work packets.
However, the model does not have to be hierarchical at all. It is just that the efficiency is not any more that great. So assume you have independent contributions to the likelihood, but all parameters are shared. In order to be able to use the parallelization trick, you would have to sub-divide the data into S subsets which form the packets of work sent to the child worker nodes. To do that you will have to copy the shared parameters S times and by that increase the auto-diff stack which is costly to do. Hence, it’s not super-optimal, but you should still benefit from it if the likelihood evaluation is costly.
In short, the user has to come up with packets of work and the map function Bob described will distribute the work. This can be applied to any model – I haven’t thought about it that way as I am almost always use some hierarchical structure.
Even a simple non-hierarchical regression will be easy to break up. And it can be broken up multiple ways. If I have a data matrix $latex x$ and a coefficient vector $latex \beta$, and lets say a simple regression $latex \log \mbox{Normal}(y \mid x \, \beta, \sigma)$, then that reduces to $latex N$ independently evaluatable log likelihood contributions, $latex \sum_n \log \mbox{Normal}(y_n \mid x_n \, \beta, \sigma)$. So break that $latex N$ down into mini-batches, ship them out, and the reduction’s just a sum at the end. Given that we’ll be able to ship the data $latex x$ out to local processors, the communication cost is the size of $latex \beta$. That can be prohibitive in the “big data” models for things like natural language processing (my old field), where $latex x$ is verys parse and $latex \beta$ is very high-dimensional. But we could probably just send back the non-zero gradients if we were clever with the mapping.
So 40 cores was two machines each with 20 cores?
Is the scaling efficiency lost beyond 20 cores due to inherent algorithm unscalability beyond twenty for this case or did the inter-machine network latency kick in to degrade performance?
I ran this on our Novartis cluster where we have infiniband which is a high-performance network system. So, yes, I think it ran on 2 machines for the 40 core run (I am not good at teasing this info out from SunGrid).
Certainly, the communication cost is critical, but look at the numbers: I get a 31x speedup on 40 cores. So not much of an efficiency loss here. I suspect that with a larger problem size the efficiency degradation may not have been that much.
Having said that, the case of ODEs is probably the best application for this as these are so expensive to evaluate. However, given the near linear speedup, I am quite optimistic that other non-ODE models will also benefit, but that remains to be tested. The good thing is that the MPI approach can parallelize from a technical perspective any Stan program.
For comparison do you have scaling results over a regular Ethernet fabric? Is the scaling latency sensitive?
This is indeed super cool. It’s a bit more general than using GPUs, becuase it’s more general and most Stan models aren’t amenable to GPU memory bandwidth or parallelization. That’s why Steve Bronder and Rok Cesnovar are starting with Cholesky factorization for GPUs — it’s quadratic memory transfer, but very parallelizable cubic operations. Then they’ll probably do matrix-matrix multiply and then maybe matrix-vector multiply when the data can be shipped out safely. Using GPUs is going to loosen some prohibitive logjams with models involving big matrix operations like large-scale simple regressions or things like Gaussian processes.
@Andrew: I’m guessing it’s the default in whatever graphing package Sebastian used. As you know from your own publications, it’s often not worth the effort taking all your advice on graphing.
@zdr: The main bottleneck is the automatic differentiation. You have to pass derivatives back along the expression graph using the chain rule and it requires the parent of each node to have its adjoint (derivative with respect to the final result) calculated before you calculate the adjoints of its children. What happens with the parallelization is that we can evaluate a bunch of the chain rule stuff remotely and just send back the result and partials (the master process knows the addresses of the operands). I don’t know what “weak scaling” is, but this will be highly parallelizable for any likelihood (or prior term for that matter) that’s parallelizable. We can use OpenMP down at the matrix computation level, but it’s harder to do that for our autodiff because of the serialization constraint on the expression graph and the fact that we (like just about everyone else with efficient autodiff) use non-threadlocal memory for the expression graph. Parallelizing over processes solves that problem without the penalty of using thread local variables in tight loops. I agree that it’d be interesting to see where communication costs dominate. As you can see, it’s well past where we’re at with 40 cores.
@Nick Menzies and @MJT: Our initial plan is to use a map function in the functional programming sense, as in the sapply() / lapply() functions in R or the map() function in Lisp. So yes, it won’t automatically apply to a model—the parallelization will have to be coded using the map function. Then the mapped operations can be parallelized, including pushing the required data out only once so only parameters need to be shared back and forth. This will really cut down on inter-process communication. Then we reduce it back into the expression graph (in the map-reduce sense).
@Rahul: Scaling is always latency sensitive, so yes. How much it’s sensitive is the real question.
@Bob The scaling results above are ‘strong scaling’, i.e. a case where the problem size stays fixed as one adds more processors. ‘Weak scaling’ is when the problem size per processor is fixed, so the problem grows linearly with the number of added computational units. (The SciNet wiki is the only weblink I could find with relevant pictures: https://wiki.scinet.utoronto.ca/wiki/index.php/Introduction_To_Performance#Weak_Scaling_Tests)
The reason I care about weak scaling is that it measures how large a problem one can handle with a given code. As I understand it, one of the major issues in Bayesian computation is how to handle so-called big data. I don’t work on MCMC codes (I work closer to computational fluid dynamics), but it seems like your computational burden grows larger as your dataset grows. So I’m curious if parallelizing in the way you described allows you to handle larger datasets. A weak scaling test would measure that, and would give some hints as to how large a dataset you could handle.
Hmmm, is parallelizability a function of the model? Could an analyst make sacrifices in the modeling in order to scale to a larger problem?
@Bob:
>>>How much it’s sensitive is the real question.<<<
Yes, that's my question. One way to find out is to compare scaling graphs for runs over standard ethernet vs a cluster having Infiniband / Myrinet etc.
Another could be to compare (say) 16-core-single-server runs vs 16-core-8-server runs and such combinations.
Just curious, what’s the reason for this internal constraint:
>>>the gradient calculation must not run in a threaded program<<<
I think elsewhere Bob said it had to do with the fact that the auto-diff code uses shared memory for the evaluation tree. It’s not clear to me if that means you couldn’t partition the tree into separate subtrees and do threaded eval of each subtree.
Afraid not—we (and pretty much everyone else using reverse-mode autodiff) use a global memory stack. Everything’s written up in our arXiv paper on Stan’s autodiff.
It is straightforward to just declare that global memory for the expression graph to be thread local. Then we can do the same thing we can do with MPI. But there’s a penalty for making the variables thread local—it was around 15 or 20% without any thread contention if I’m remembering correctly from when I tried it several years ago. This is all going to be much easier to test with the built-in threading library in C++11.
As it is, with many models I have worked on involving large numbers of parameters (several thousand), the big bottleneck is warmup. Getting into the typical set takes 20 to 50 iterations, each iteration needs to use a small step size, and therefore uses a treedepth of 10 to 15, meaning 1000 to 30,000 timesteps of hamiltonian integration. Being able to parallelize that so that I get into the typical set quickly (in wall time) would be a huge win.
In fact, only after getting into the typical set would I like to then using all the adaptation info fork off 4 separate chains from 4 randomly chosen starting points from within the typical set.
I recognize this has issues with convergence detection, but it still seems better than doing all that warmup in separate processes rather than parallelizing the warmup. Tradeoffs I guess.
Andrew always wants us to run multiple replicates of the entire process to test convergence. Easy if you run simple models like he tends to fit, but more challenging if you don’t have access to (or patience to configure and monitor) lots of machines.
Branching after you think you get into the typical set would be better than nothing. But the real problem is that you not only need to get into the typical set, you need to stay there long enough to estimate the (co)variance.
There’s nothing stopping us from parallelizing warmup itself. It doesn’t even have to be a Markov chain.
In these kind of cases, I always wind up with 1 on the mass matrix anyway, it doesn’t seem to do any adaptation, perhaps because I give it such small warmups, so I wind up running with tiny step sizes, and each sample takes 1 to 10 minutes, Just getting me into the typical set is taking long enough. If I could parallelize that I’d be happy. Also, I’d like to tell stan to run until you get into the typical set, and then give me N markov draws, let it decide when it’s in the typical set, something like looking at local trend line of the lp__ and waiting until it becomes near zero, then switch automatically to sampling, so instead of telling it iter=100, warmup=50, i’d just say markov.samps=50 and let it run warmup until it’s in the typical set and then do 50 markov samples.
This is awesome! By making big models faster and more efficient, it’ll open up huge new areas of applied research. Do you have a tentative timeline as-to when we should expect to see this in the main Stan distribution?